Find problems which are often encountered when using MASON
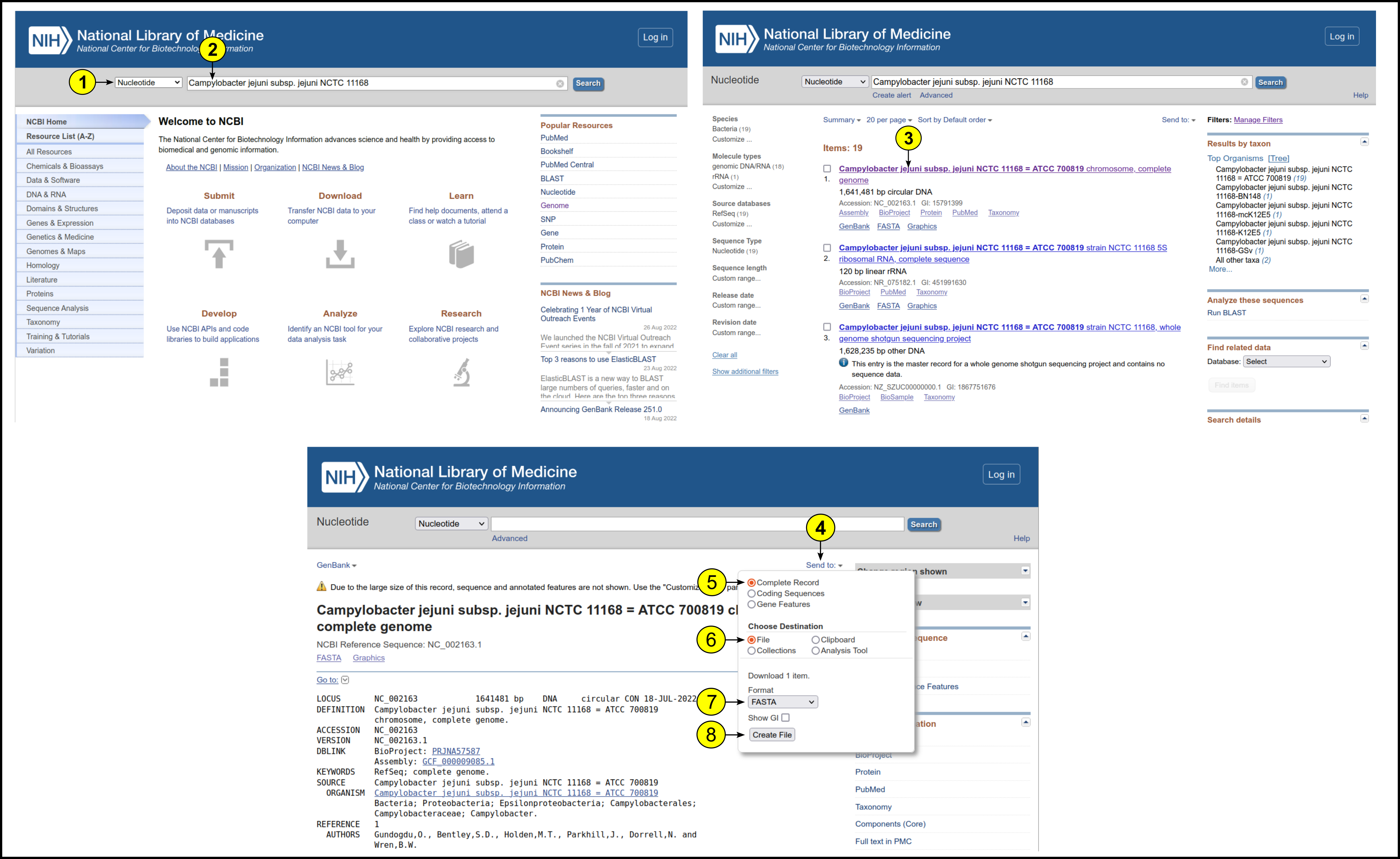
How to find and download a genome/annotation file in NCBI?
To upload a custom organism to MASON, the genome sequence and annotation files must be provided in FASTA and GFF format, respectively. Below we describe an example explaining how to find and download these files for the pathogenic bacterium Campylobacter jejuni:
- Open the NCBI website and select “Nucleotide” in the dropdown menu
- Add the name of your target organism (here, “Campylobacter jejuni subsp. jejuni NCTC 11168”) and click the search button
- Once the query results are shown, select the complete genome of your target organism and click on it
- Click “Send to” to download available files for the organism
- Select the “Complete Record button”
- Click on the “File” button
- Select “FASTA” from the dropdown menu
- Click “Create File” to download the FASTA file of the complete genome
- Repeat steps 4-8, but select “GFF3” instead of “FASTA” at step 7 to download the annotation file in GFF format
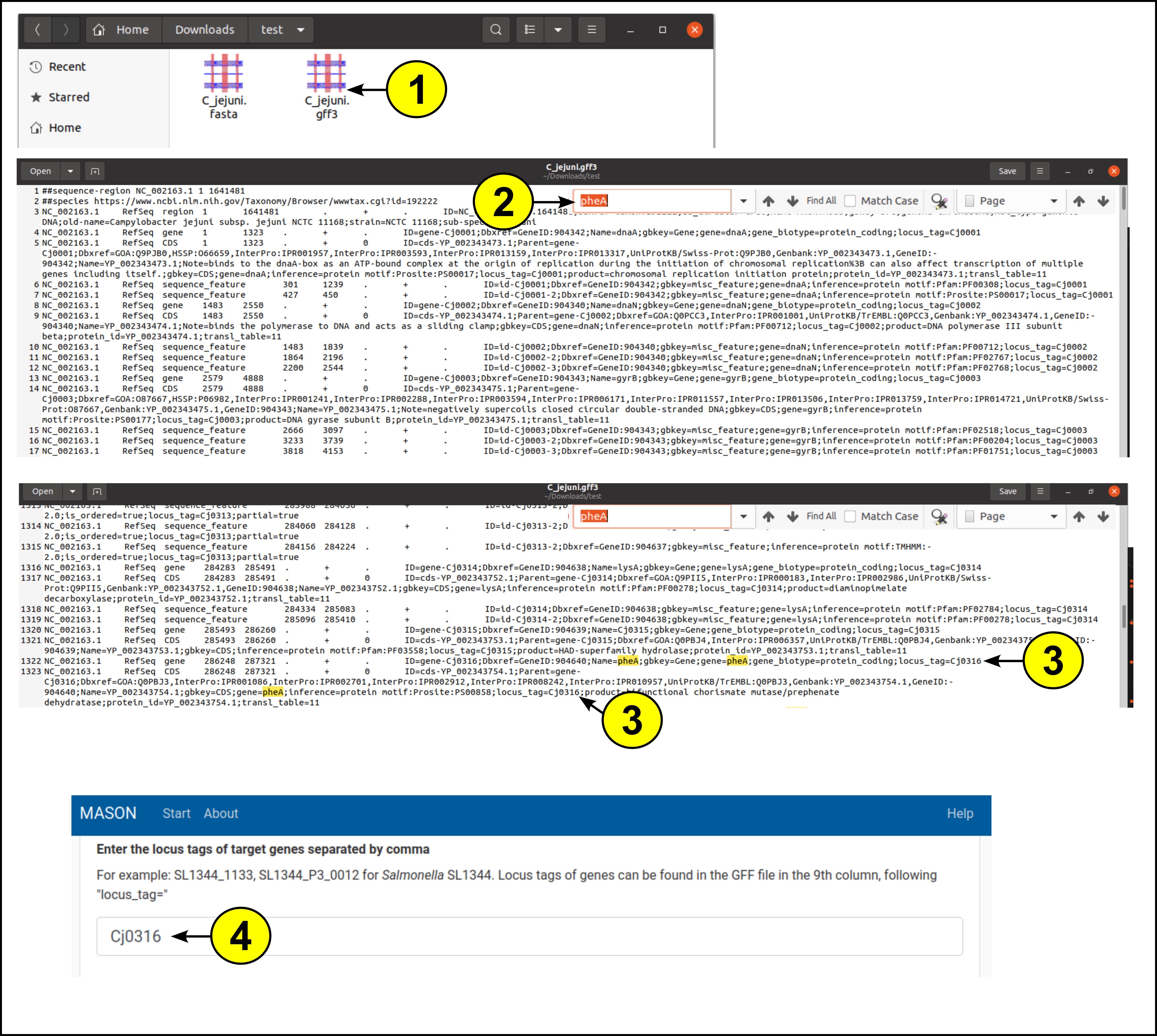
How to find the locus tag of my selected target gene in the target organism?
MASON requires locus tags as identifiers for target genes, because they are more consistent across species than gene names. Below we describe an example explaining how to find the locus tag of the essential gene “pheA” in Campylobacter jejuni:
- Open the GFF annotation file in your favourite text editor
- Use the search and find utility of your text editor. Often, CTRL + F opens up the “Find” tool. Now enter the name of your target gene (here, “pheA”) and press ENTER
- In the 9th column of the line, information of the gene is stored. Find the “locus_tag=” field and copy the locus tag
- Add the copied locus tag to MASON as a target gene
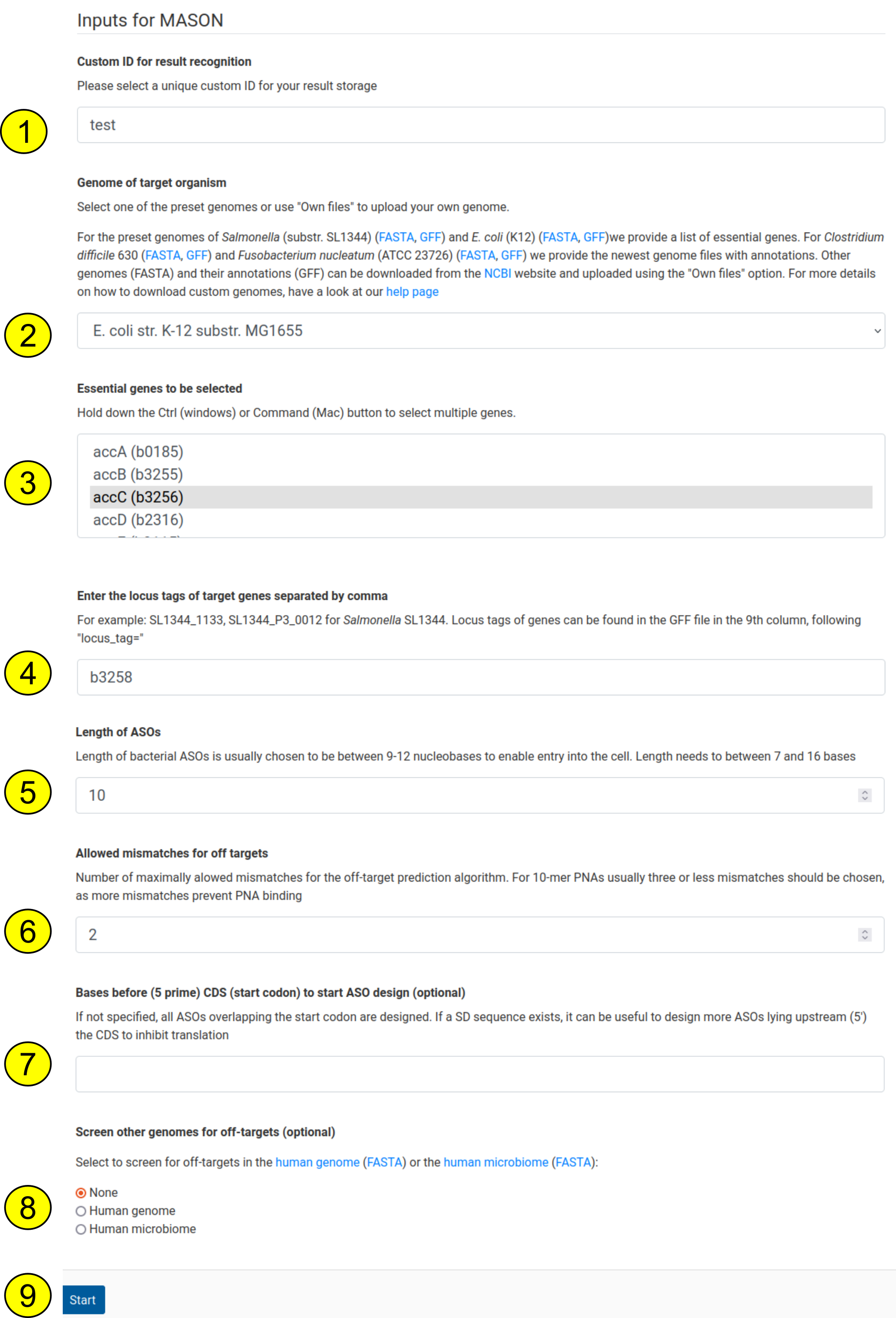
How to fill in the start form?
Below each start form is a short description of what to fill in. We show an example on a case in which a user uses MASON to find ASO sequences for one essential gene (accC) and another, non-essential gene (b3256) in E. coli K12:
- The custom name can be any name, here we use “test”
- We use the pre-selectable genome of E. coli K12. Likewise, other genomes can be selected or a custom genome can be uploaded, see the other help page for information on this
- We select the essential gene “accC”
- We additionally select another gene, with the locus tag “b3258”. This is optional and can be left out if already a gene in the dropdown menu was selected
- The length of the ASOs is chosen to be 10 to get 10mer ASOs
- 2 mismatches are allowed to be inside the ASOs to be considered a mismatch
- We want to design only sequences targeting the start codon. Therefore, this option is left empty
- No other genome is selected for off-target screening. Optionally, we could have added the human genome or the microbiome to be screened for off-targets
- The start button can now be clicked to start the MASON process
How does the machine-learning option work?
If the "Use machine-learning (random forest) model to predict MICs" box is ticked, MASON feeds each designed ASO into a random forest regressor and returns a predicted minimum inhibitory concentration (MIC) along with a relative rank.
- The model was trained on an internal, unpublished dataset of ~586 PNAs targeting the essential genome of uropathogenic E. coli str. 536 (UPEC 536). Each PNA in the training set was assayed for its MIC against UPEC 536.
- Input features include PNA sequence properties (Tm, % self-complementary bases, purine percentage), the number of off-targets in TIR regions of UPEC 536 (1 mismatch), the codon-adaptation index (CAI) of the target gene, and the minimum free energy (MFE) of the target's translation initiation region.
- Predicted MIC values are reported in µM. Because the model was trained on UPEC 536 data, they should not be interpreted as absolute MICs in other organisms — instead, use the MIC_rank column to compare ASOs relative to each other.
What is a SHAP value?
SHAP (SHapley Additive exPlanations) values explain individual predictions by attributing
each feature a contribution that pushes the model output above or below the average
prediction (the "base value"). They are based on game-theoretic Shapley values and are
computed here with the
shap Python library
applied to the random forest MIC predictor.
For every designed ASO we generate a force plot which can be opened by hovering over the SHAP link in the MIC_predicted column of the result table. In the plot:
- the bold value on the axis is the model's predicted MIC for that ASO,
- the "base value" is the average predicted MIC across the training set,
- orange bars push the prediction up (worse predicted MIC), dark blue bars push it down (better predicted MIC), and the size of the bar reflects how strongly that feature moved the prediction.
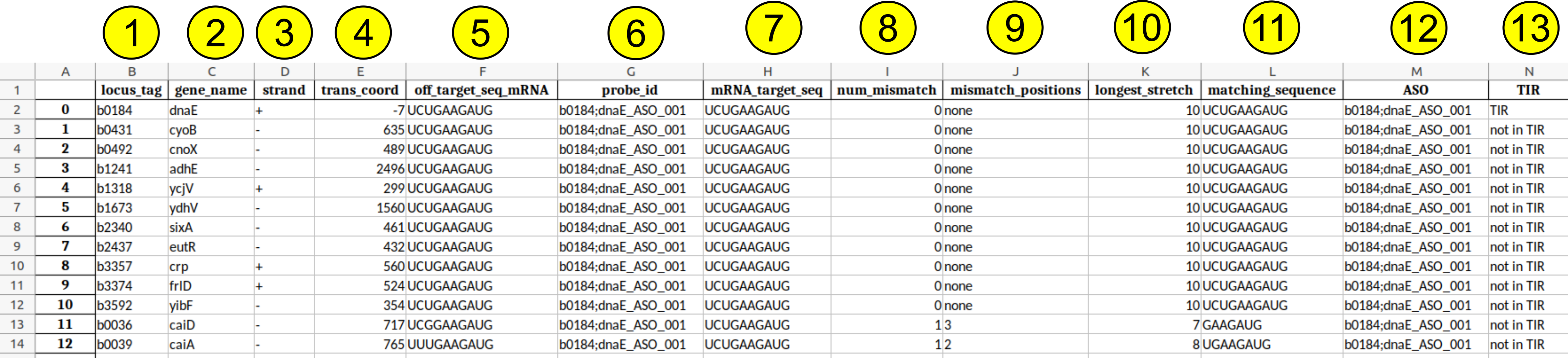
How to interpret the off-target output table?
Below each output column is a short description of what it means. Each row denotes an off-target match with a specific gene of a respective PNA.
- locus_tag: Denotes locus tag of off-target gene
- gene_name: Denotes gene name of off-target gene
- strand: Denotes strand of off-target match (target mRNA)
- trans_coord: Denotes the location of off-target match. I.e. first matching base respective to start codon of off-target gene
- off_target_seq_mRNA: Denotes sequence of mRNA of organism that is subject to an off-target effect
- probe_id: Unique identifier of off-target. Consists of locus tag (target gene), gene name (target gene) and ASO name
- mRNA_target_seq: Denotes targeted sequence of ASO. I.e. the target mRNA if there are no mismatches
- num_mismatch: Number of mismatches of off-target
- mismatch_positions: Positions of mismatches
- longest_stretch: length of longest matching stretch without a mismatch
- matching_sequence: longest matching stretch without a mismatch (ASO-mRNA, mRNA sequence shown)
- ASO: Unique name of the ASO
- TIR: Whether the off-target is within the translation initiation region of a gene
If there are further errors, requests for help, or suggestions for additional features, please email to jakobjung@tutanota.com and/or open an issue on the github page of MASON: github.com/BarquistLab/mason/issues
Research institutes & networks



